
50條負面清單發(fā)布
近日,山東省藥品監(jiān)督管理局發(fā)布《醫(yī)療器械經營企業(yè)、使用單位質量安全主體責任清單和負面清單的通知》(以下簡稱《通知》)。
《通知》稱,山東省藥監(jiān)局依據《醫(yī)療器械監(jiān)督管理條例》《醫(yī)療器械經營監(jiān)督管理辦法》《醫(yī)療器械使用質量監(jiān)督管理辦法》等,研究制定了醫(yī)療器械經營企業(yè)、使用單位質量安全主體責任清單和負面清單。
具體如下:
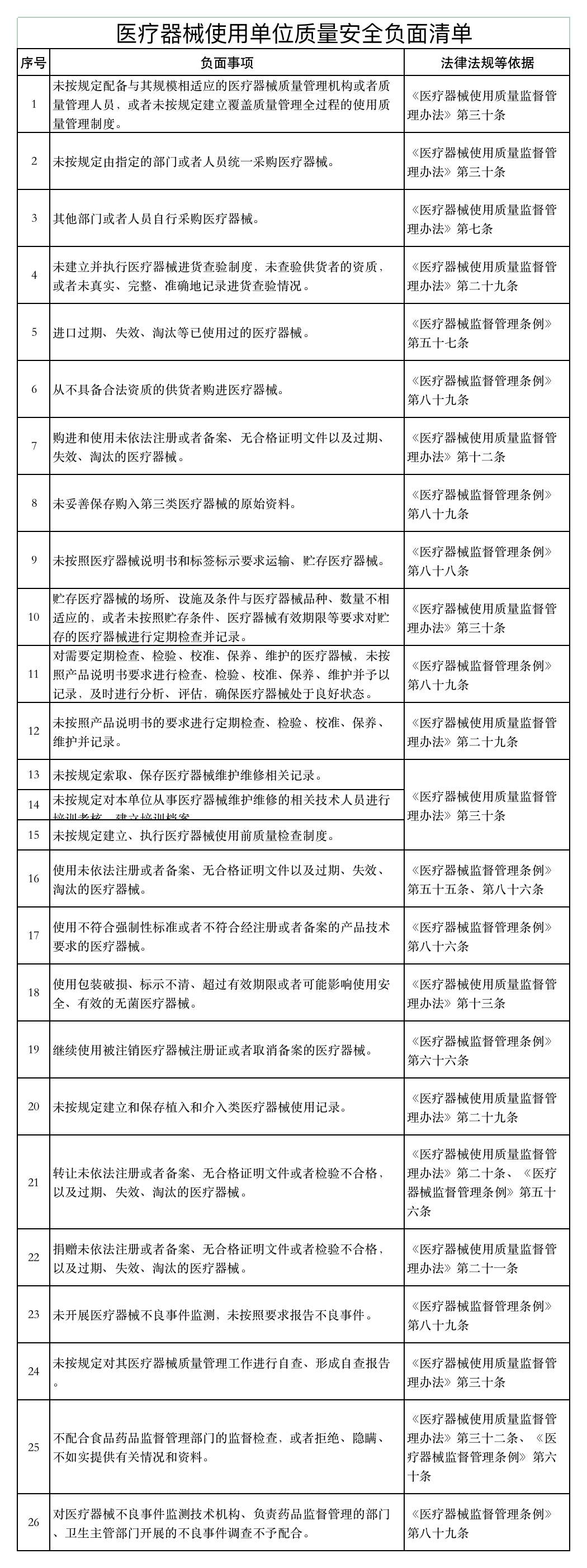
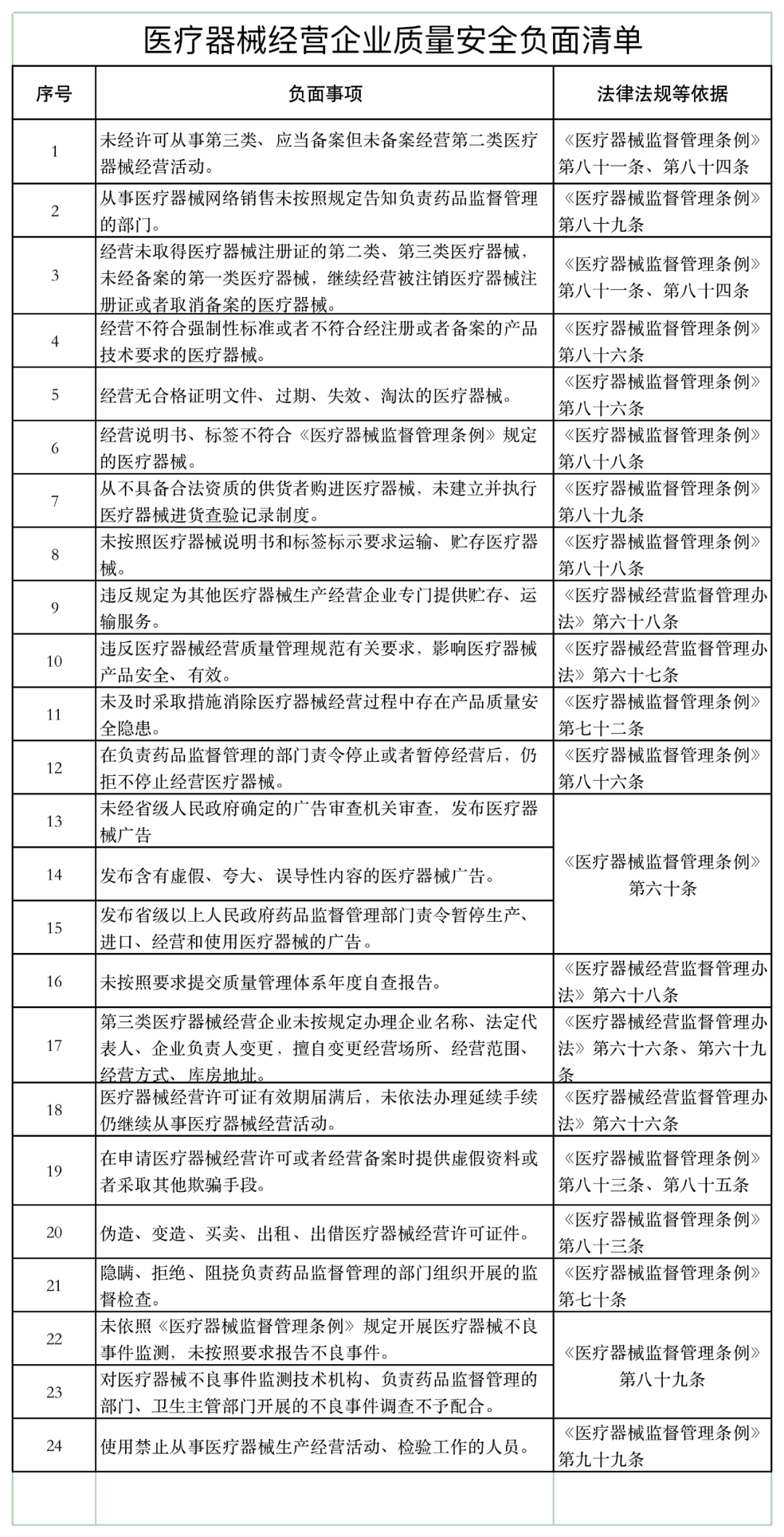
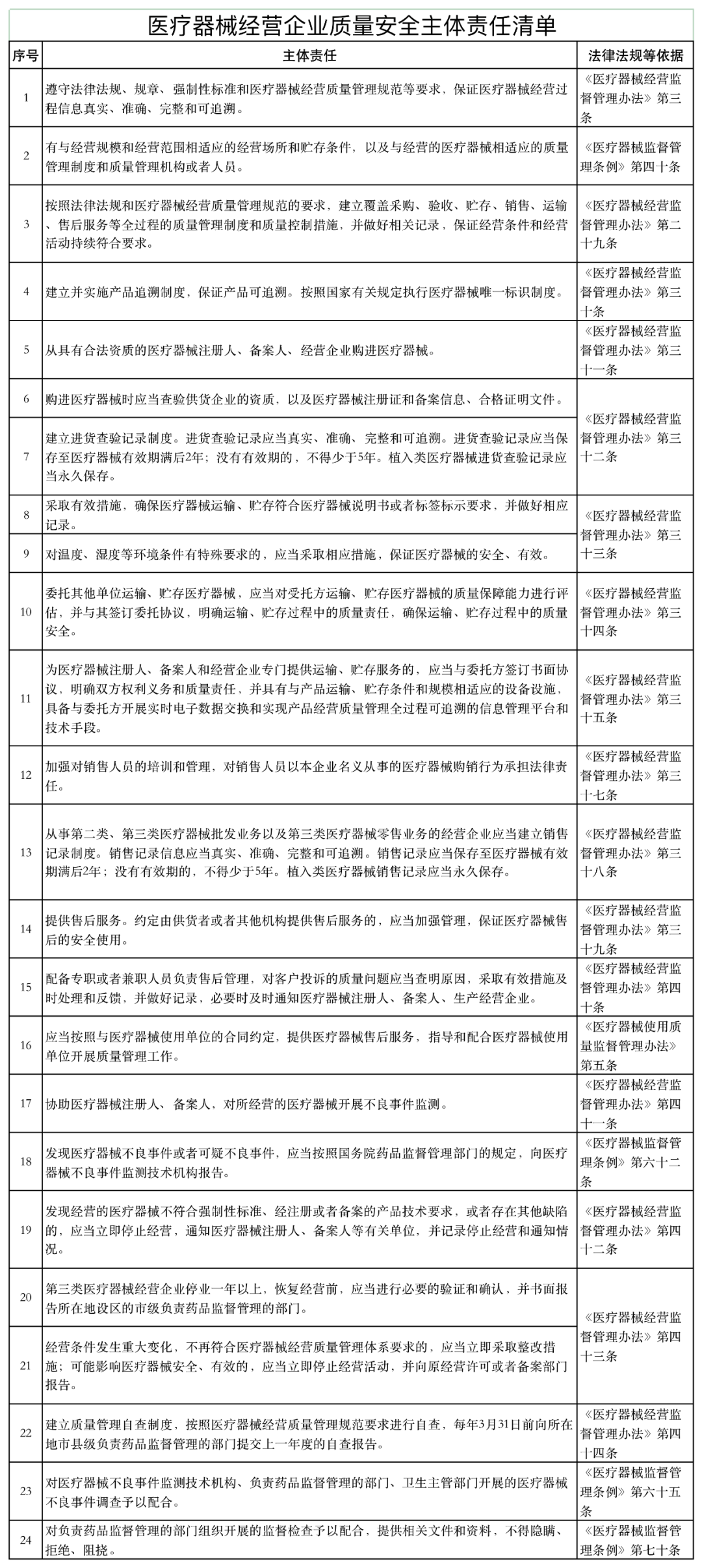
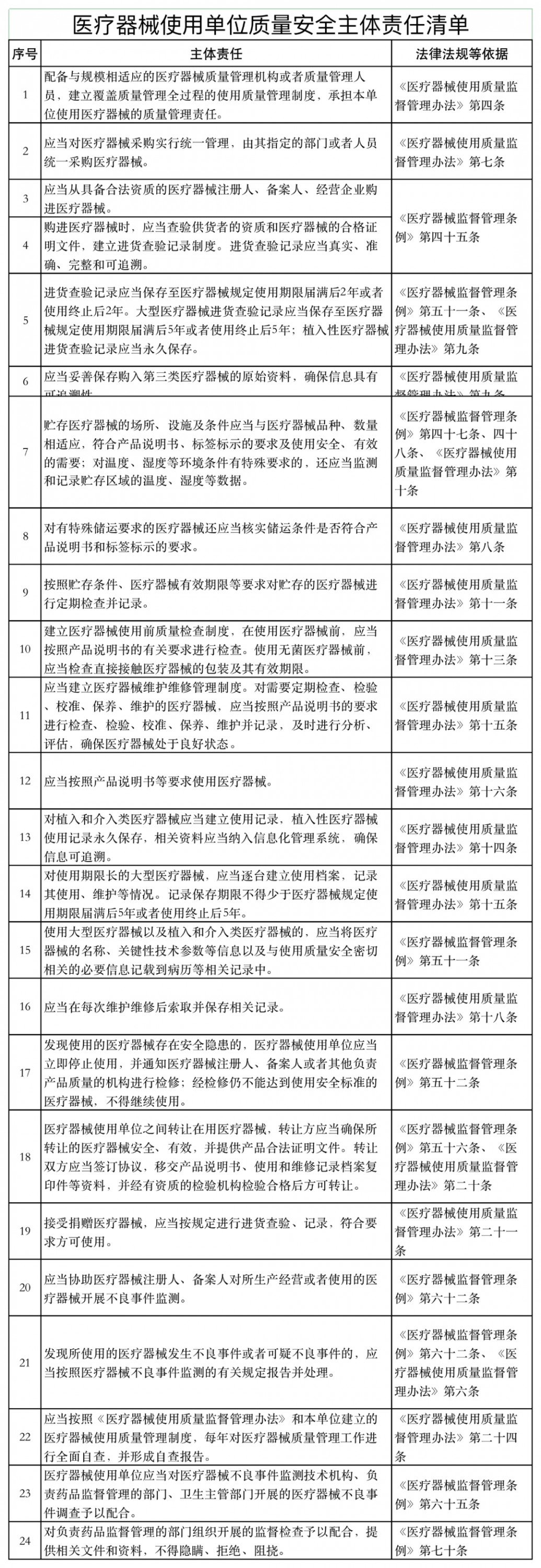
醫(yī)療器械經營監(jiān)管大調整
隨著新版《醫(yī)療器械生產監(jiān)督管理辦法》《醫(yī)療器械經營監(jiān)督管理辦法》的落地執(zhí)行,醫(yī)療器械企業(yè)的經營和監(jiān)管力度和措施都提升到新的緯度。
國家指出,兩個辦法嚴格貫徹落實“四個最嚴”要求。落實《醫(yī)療器械監(jiān)督管理條例》規(guī)定,全面落實醫(yī)療器械注冊人備案人制度,優(yōu)化行政許可辦理流程,強化監(jiān)督檢查措施,完善監(jiān)督檢查手段,強化企業(yè)主體責任,并進一步加大對違法行為的處罰力度。
《經營辦法》一方面進一步強化了企業(yè)質量責任。要求企業(yè)從事醫(yī)療器械經營,應當按照法律法規(guī)和醫(yī)療器械經營質量管理規(guī)范的要求,建立覆蓋采購、驗收、貯存、銷售、運輸、售后服務等全過程的質量管理制度和質量控制措施,并做好相關記錄,保證經營條件和經營活動持續(xù)符合要求。
同時,醫(yī)療器械經營企業(yè)應當建立質量管理自查制度,按照醫(yī)療器械經營質量管理規(guī)范要求進行自查,每年3月31日前向所在地市縣級負責藥品監(jiān)督管理的部門提交上一年度的自查報告。
另一方面更加注重經營全過程的質量管理。
一是要求醫(yī)療器械經營企業(yè)應當從具有合法資質的醫(yī)療器械注冊人、備案人、經營企業(yè)購進醫(yī)療器械,嚴格控制采購和銷售環(huán)節(jié)的資質審核,確保醫(yī)療器械的合法流通。
二是重點突出了進貨查驗、銷售環(huán)節(jié)的記錄要求,保證產品的可追溯,并應當按照國家有關規(guī)定執(zhí)行醫(yī)療器械唯一標識制度。
三是強調了對低溫、冷藏醫(yī)療器械的儲運要求,確保產品運輸質量。四是對經營企業(yè)的售后服務提出要求,確保產品的使用安全。
新《經營辦法》通過如下措施強化監(jiān)管:一是實施分類分級管理。藥品監(jiān)督管理部門根據醫(yī)療器械經營企業(yè)質量管理和所經營醫(yī)療器械產品的風險程度,實施分類分級管理并動態(tài)調整。
二是制定年度檢查計劃。設區(qū)的市級、縣級負責藥品監(jiān)督管理的部門應當制定年度檢查計劃,明確監(jiān)管重點、檢查頻次和覆蓋范圍并組織實施。三是進行延伸檢查。
藥品監(jiān)督管理部門根據醫(yī)療器械質量安全風險防控需要,可以對為醫(yī)療器械經營活動提供產品或者服務的其他相關單位和個人進行延伸檢查。四是風險會商研判。藥品監(jiān)督管理部門應當根據監(jiān)督檢查、產品抽檢、不良事件監(jiān)測、投訴舉報、行政處罰等情況,定期開展風險會商研判,做好醫(yī)療器械質量安全隱患排查和防控處置工作。
信用檔案建設。設區(qū)的市級負責藥品監(jiān)督管理的部門應當建立并及時更新轄區(qū)內醫(yī)療器械經營企業(yè)信用檔案。
《生產辦法》方面,在現有的醫(yī)療器械生產許可和備案、監(jiān)督檢查、責任約談等監(jiān)管方式方法的基礎上,從四個方面進一步豐富完善監(jiān)管手段。
一是建立醫(yī)療器械報告制度。規(guī)定年度報告、生產產品品種報告、生產條件變化報告和重新生產報告四種報告形式,以便監(jiān)管部門及時掌握企業(yè)的生產狀況,有針對性地采取監(jiān)管措施。
二是進一步完善監(jiān)督檢查方式方法。明確監(jiān)督檢查、重點檢查、跟蹤檢查、有因檢查和專項檢查等多種監(jiān)督檢查形式,并對有因檢查和跟蹤檢查的內容和方式作出具體規(guī)定。
三是細化明確信息公開和責任約談制度。藥品監(jiān)督管理部門依法及時公開醫(yī)療器械生產許可、備案、監(jiān)督檢查、行政處罰等信息,方便公眾查詢,接受社會監(jiān)督。醫(yī)療器械注冊人、備案人、受托生產企業(yè)對存在的醫(yī)療器械質量安全風險,未采取有效措施消除的,藥品監(jiān)督管理部門可以對醫(yī)療器械注冊人、備案人、受托生產企業(yè)的法定代表人或者企業(yè)負責人進行責任約談。
委托生產如何開展?
醫(yī)療器械注冊人、備案人委托生產的,應當對受托方的質量保證能力和風險管理能力進行評估,按照國家藥監(jiān)局制定的委托生產質量協議指南要求,與其簽訂質量協議以及委托協議,監(jiān)督受托方履行有關協議約定的義務;
受托生產企業(yè)應當按照法律、法規(guī)、規(guī)章、醫(yī)療器械生產質量管理規(guī)范、強制性標準、產品技術要求、委托生產質量協議等要求組織生產,對生產行為負責,并接受醫(yī)療器械注冊人、備案人的監(jiān)督。
受托生產企業(yè)應當向原生產許可或者生產備案部門報告增加生產的產品品種情況,并提供委托方、受托生產產品、受托期限等信息;
增加生產產品涉及生產條件變化,可能影響產品安全、有效的,應當在增加生產產品30個工作日前向原生產許可部門報告,原生產許可部門應當及時開展現場核查。屬于許可事項變化的,應當按照規(guī)定辦理相關許可變更。
醫(yī)療器械注冊人、備案人應當負責產品上市放行,建立產品上市放行規(guī)程,明確放行標準、條件,并對醫(yī)療器械生產過程記錄和質量檢驗結果進行審核,符合標準和條件的,經授權的放行人員簽字后方可上市。
委托生產的,醫(yī)療器械注冊人、備案人還應當對受托生產企業(yè)的生產放行文件進行審核。
受托生產企業(yè)應當建立生產放行規(guī)程,明確生產放行的標準、條件,確認符合標準、條件的,方可出廠。不符合法律、法規(guī)、規(guī)章、強制性標準以及經注冊或者備案的產品技術要求的,不得放行出廠和上市。醫(yī)療器械注冊人、備案人不得委托受托生產企業(yè)進行上市放行。
落實生產產品品種報告制度。醫(yī)療器械生產企業(yè)應當向藥品監(jiān)督管理部門報告所生產的產品品種情況。
增加生產產品品種的,應當向原生產許可或者生產備案部門報告,涉及委托生產的,還應當提供委托方、受托生產產品、受托期限等信息。醫(yī)療器械生產企業(yè)增加生產產品涉及生產條件變化,可能影響產品安全、有效的,應當在增加生產產品30個工作日前向原生產許可部門報告,原生產許可部門應當及時開展現場核查。屬于許可事項變化的,應當按照規(guī)定辦理相關許可變更。


